It would seem reasonable to believe Pfizer and its corporate associates may be selectively reducing the associated risk of mRNA gene therapy vaccination for their employees and their employees families by providing them, through an employee vaccination program, with vaccine batches produced using a separate manufacturing process to the commercial scale one used to supply general populations.
Evidence for this is as follows:
- It is known that the mRNA gene therapy vaccine solution can be produced using two substantively different processes, a clinical process and a commercial process.
- It is known that the European Medicines Agency(EMA) expressed concerns with Pfizer over discrepancies between the clinical and commercial batches, pertinent to the safety and efficacy of the mRNA gene therapy vaccine solutions produced, and that commercial batches have a higher potential for risk than clinical batches.
- It is known that Pfizer and Pfizer corporate associates vaccinate their employees and their employee's families with batches distinct and separate from the commercial ones given to general populations.
- It is reasonable to believe that the data provided by the United States post market surveillance system, VAERS, suggests a profoundly uneven distribution of reported incidents of adverse events(AE) and death across batches of the mRNA gene therapy vaccine solution, indicating some batches are significantly more risky than others.
- It is known, that at least in Australia, Pfizer employee batches are not tested by the country's regulatory agency which tests all commercial batches, and those batches marked for allocation to Pfizer employees have undergone odd modifications as to their regulatory status within the batch registry database.
1. Commercial and Clinical Batch Processes
There are two substantively different production processes used in the manufacture of the Pfizer-BioNTech Gene Therapy Vaccine solution.
The clinical batch process was used to produce the solution administered in the clinical trials, data from which formed the safety and efficacy profile used to authorise use of another commercial batch process for mass production of the solution given to the general public.
Concerns arising from were one of the “Major Objections” intially raised with Pfizer by the European Medicines Agency during the authorisation process, as outlined in their assessment report for the product:
Manufacturing process development
Process development changes were adequately summarised. Two active substance processes have been used during the development history; Process 1 (clinical trial material) and Process 2 (commercial process). Details about process differences, justification for making changes, and results from a comparability study are provided. The major changes between active substance process versions were described in the dossier.
Batch analysis results showing comparability between non-clinical and clinical batches are provided. Additional characterization of product-related species and their relation to final product specifications will be provided as a specific obligation.
Electropherograms were presented demonstrating similarities in the peak pattern of RNA species, but some differences between Process 1 and 2 were also noted. It can therefore not be concluded that identical species are obtained by the processes. It is likely that the fragmented species will not result in expressed proteins, due to their expected poor stability and poor translational efficiency (see below). However, the lack of experimental data on the truncated RNA and expressed proteins does not permit a definitive conclusion and needs further characterisation. Therefore, additional characterisation data remain to be provided as a specific obligation (SO1).
– Comirnaty(Pfizer BioNTech) COVID-19 mRNA vaccine Assessment Report, page 18
Efficacy, safety and immunogenicity was demonstrated using clinical batches of vaccine from Process 1. The commercial batches are produced using a different process (Process 2), and the comparability of these processes relies on demonstration of comparable biological, chemical and physical characteristics of the active substance and finished product.
– Comirnaty(Pfizer BioNTech) COVID-19 mRNA vaccine Assessment Report, page 34
Data demonstrate the presence of significant amounts of truncated/modified forms of mRNA at somewhat higher levels in the batches manufactured with the commercial process as compared to material used in clinical trials. These forms are poorly characterised, and the limited data provided for protein expression do not fully address the uncertainties relating to the risk of translating proteins/peptides other than the intended spike protein.
– Comirnaty(Pfizer BioNTech) COVID-19 mRNA vaccine Assessment Report, page 35
2. Discrepancies between Commercial and Clinical Batches
These concerns raised with the divergent production processes were further amplified in a subsequent leak of internal EMA documentation and emails to academic journals as a result of a cyber attack launched against the agency during December 2020.
The BMJ has reviewed the documents, which show that regulators had major concerns over unexpectedly low quantities of intact mRNA in batches of the vaccine developed for commercial production.
EMA scientists tasked with ensuring manufacturing quality—the chemistry, manufacturing, and control aspects of Pfizer’s submission to the EMA—worried about “truncated and modified mRNA species present in the finished product.” Among the many files leaked to The BMJ, an email dated 23 November by a high ranking EMA official outlined a raft of issues. In short, commercial manufacturing was not producing vaccines to the specifications expected, and regulators were unsure of the implications. EMA responded by filing two “major objections” with Pfizer, along with a host of other questions it wanted addressed.
The email identified “a significant difference in % RNA integrity/truncated species” between the clinical batches and proposed commercial batches—from around 78% to 55%. The root cause was unknown and the impact of this loss of RNA integrity on safety and efficacy of the vaccine was “yet to be defined,” the email said.
Ultimately, on 21 December, EMA authorised Pfizer-BioNTech’s vaccine. The agency’s public assessment report, a technical document published on its website, noted, “the quality of this medicinal product, submitted in the emergency context of the current (covid-19) pandemic, is considered to be sufficiently consistent and acceptable.”
It’s unclear how the agency’s concerns were satisfied. According to one of the leaked emails dated 25 November, positive news had come from an undisclosed source in the US: “The latest lots indicate that % intact RNA are back at around 70-75%, which leaves us cautiously optimistic that additional data could address the issue,” the email said.”
– 'The EMA covid-19 data leak, and what it tells us about mRNA instability', Serena Tinari, British Medical Journal., archive link
3. BioNTech Employee Vaccination Program Does Not Use Commercially Produced Batches
In a December 23rd interview, BioNTech CEO Ugur Sahin states neither himself nor his employees had yet been vaccinated, but they planned to “make an extra batch independent from the European contingent” available to themselves and corporate partners. The direct quote can be found in the below video.
The employee specific program he proposed was then instituted in January with further details of it announced in a company press release:
The additional small quantities of vaccine used for this purpose are separate and distinct from those committed under the supply agreements entered in to with the European Commission and other governments, and will not impact BioNTech’s ability to meet its commitments under these supply agreements.
– Statement on Voluntary COVID-19 Vaccination for BioNTech Employees and Suppliers to Ensure Undisrupted Manufacturing and Distribution of COVID-19 Vaccine Doses, archive link
So the BioNTech employee and associate mRNA gene therapy vaccine solution was produced in small quantity batches "separate and distinct" from those "committed under the supply agreements" as to not "impact BioNTech’s ability to meet its commitments under these supply agreements". In other words, BioNTech did not use the commercial batch manufacturing process for itself and instead used the clinical batch manufacturing process. And it did this ostencibly to meet its commercial supply commitments.
4. Uneven Distribution of Reported AE and Death Across Batches of mRNA Gene Therapy Vaccine Solution
Data analysis using the VAERS database, the authoritative system used in the United States to report vaccine induced adverse events and deaths through post-market surveillance, would appear to show an uneven distribution of both reported deaths and AE across vaccine batch numbers for both the Moderna and Pfizer mRNA gene therapy vaccines. I have not verified the following analysis, but they are done using publicly avaliable data.
100% of Covid-19 Vaccine Deaths were caused by just 5% of the batches produced according to official Government data, archive link
100% of Covid-19 Vaccine Permanent Disabilities and Deaths among Children were caused by just 6% of the batches produced according to official Government data, archive link
A third source set out to fact-check the analysis done in the dailyexpose article, and ended up corroborating its findings.
“There is an article floating around from The Expose that makes an explosive claim: There is a wildly statistically-significant skew in the death rate from Covid-19 vaccines by lot number.
What originally got my attention was the tinfoil hat crowd screaming about lots being intentionally distributed to certain people to kill them -- in other words certain Covid-19 vaccine lots were for all intents and purposes poisoned. That was wildly unlikely so I set out to disprove it and apply some broom handles to the tinfoil hatters heads. What I found, however, was both interesting and deeply disturbing.
. . .
The outcome distribution isn't "sort of close" when most of the lots have a single-digit number of associated deaths.
Isn't it also interesting that when one removes the "dead" flag the same sort of correlation shows up? That is, there are plenty of lots with nearly nothing reported against them. For Moderna within the first page of results (~85 lots) there is more than a three times difference in total adverse events. The worst lot, 039K20A with 87 deaths, is not only worst for deaths; it also has more than 4,000 total adverse event reports against it. For context if you drill down a couple hundred entries in that report the number of total adverse events against another lot, 025C21A number 417 with five deaths.
Are you really going to try to tell me that a mass-produced and distributed jab has a roughly ten times adverse event rate between two lots and seventeen times the death rate between the same two, you can't explain it by "older people getting one lot and not the other" and this is not a screaming indication that something that cannot be explained as random chance has occurred?”
– 'Uh, That's Not A Conspiracy Theory, Karl Denninger, The Market Ticker, archive link
5. Irregularities in the Australian Batch Registry for Vaccine Batches Allocated to The Pfizer Employee Vaccination Program
In Australia Pfizer(and assumably their associates) employees and their families have been allocated batches of their mRNA gene therapy solution exclusive from commercial batches administered to the general population. Both the commercial batches and the Pfizer employee batches are distinctly identified in the batch registry kept by the Therapeutic Goods Administration(TGA), which is the Australian equivalent of the American FDA or European Union EMA.
Out of a total of 62 Pfizer batches registered in Australia at present, 8 of them are allocated for the Pfizer employee vaccination program. These eight are:
- FF0884* 12/8/2021
- FA4598* 30/7/2021
- FE3064* 25/6/2021
- FA7338* 22/6/2021
- FA7812* 22/6/2021
- FC8736* 22/6/2021
- FC3558* 21/6/2021
- FD0927* 7/6/2021
According to their website, Pfizer Australia has 1,700 employees. Even accounting for family and corporate partners, it remains a little odd that 15% of all Pfizer batches registered in Australia would be allocated for use in the Pfizer Employee Program. Though it turns out they were, only some of the time.
These eight batches, unlike the other 54 commercial batches, are not tested by the TGA as are all commercial batches, as they instead recognise the testing certification done by the European Union's Official Control Authority Batch Release(OCABR).
Pathway one uses overseas certification as evidence that the batch has already undergone independent testing and assessment by a recognised National Control Laboratory, such as the in Europe.
The OCABR process involves assessment of manufacturing documentation (summary protocol review) and laboratory testing for potency, identity and appearance based on guidelines for specific COVID-19 vaccine types (e.g. RNA, viral vector, and inactivated vaccines).
When the Sponsor provides evidence that the batch supplied in Australia has passed OCABR testing, the vaccine can be released without the TGA conducting a manufacturing protocol assessment or (potentially) laboratory testing.
The Sponsor must still supply samples, batch details and evidence of the maintenance of adequate shipping conditions for the batch under this pathway.
OCABR certificates are unlikely to be available for all COVID-19 vaccine batches as their availability depends on many factors, including the final global distribution of a batch.
The status of these eight batches, as reflected in the TGA batch registry system, have undergone some irregularities throughout the course of 2021. The discovery of these irregularities were made by someone conducting an independent audit of vaccine supply to the Australian government. This audit can be found here, though it isn't pertinent to the issue at hand:
Covid Supply Background Notes Covid-19 Supply AuditAs of August 10th 2021, the TGA batch registry had five batches allocated to the Pfizer employee vaccination program.
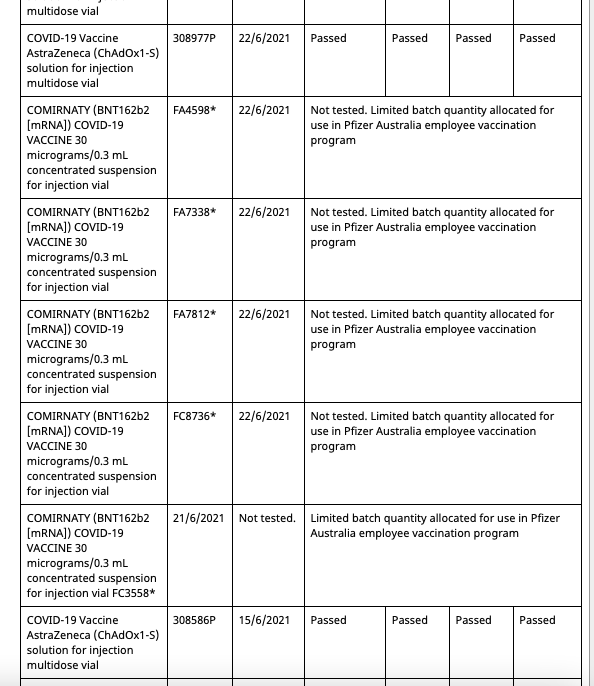
Take note of three things:
- All five entries are distinctly marked as “limited batch quantity allocated for use in Pfizer Australia employee vaccination program.”
- Batch FA4598 has an associated date of 22/6/2021
- The fifth batch at the bottom has no batch number entered, due to what would appear to be erroneous data entry.
Then on August 11th 2021, the batch registry is updated, adding a second entry for batch FA4598, but with an associated date of 30/7/2021 and status of 'Not tested, additional shipment'.
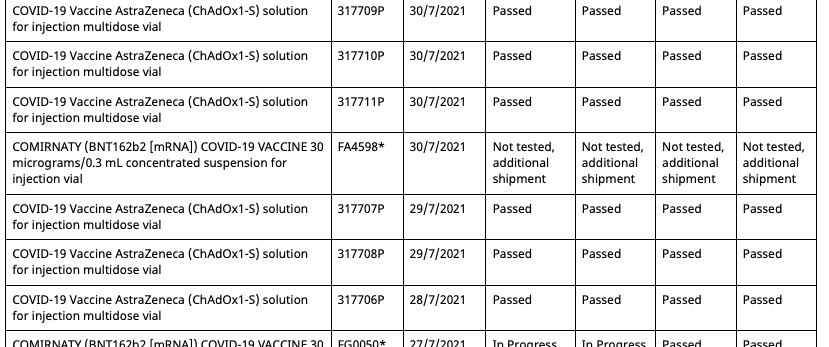
So now there are two entries in the registry for batch FA4598, one allocated to the Pfizer employee vaccination program and the other unallocated and untested. However, this duplicate batch entry shares the same OCABR certificate. Meaning that even though it is marked as untested by the TGA, it doesn't need to be tested to be allocated, and now be done so outside of the Pfizer employee vaccination program.
Then on the 19th of August the batch registry is updated once again and three key changes are made:
- The data entry error of the fifth batch has now been resolved and given the batch number FC3558
- The status of the five batches have now been changed from 'limited batch quantity allocated for use in Pfizer Australia employee vaccination program.' to 'Not tested, additional shipment'
- But there aren't five batches anymore, as batch FA4598 with the associated date of 22/6/2021 is now missing, leaving only its duplicate with the associated date 30/7/2021.
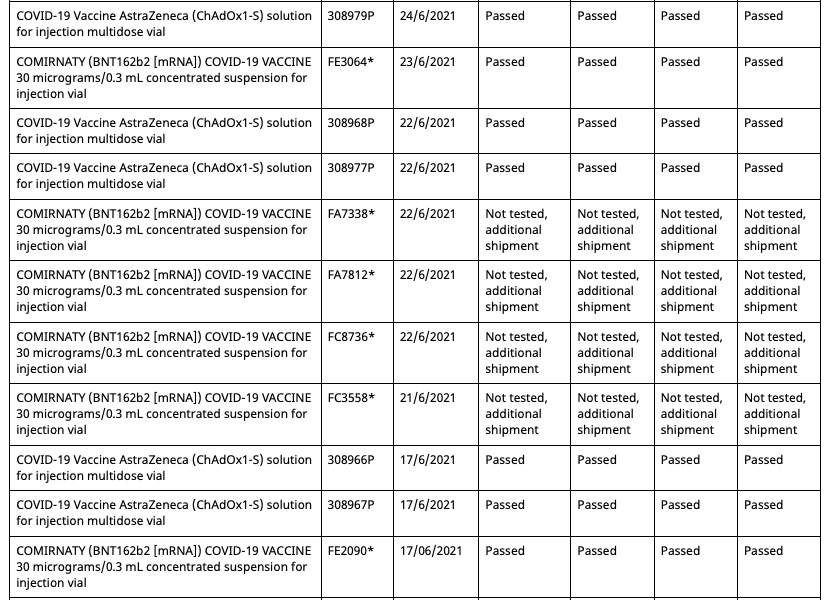
So now all Pfizer vaccination program batches can be allocated outside of the program, and have OCABR certificates so they don't need to be tested by TGA. And this remains the case until September 21st, when the following changes are made:
- The four batches from the Pfizer employee program which had their status changed to 'Not tested, additional shipment' on August 19th have had it changed back to 'Limited batch quantity allocated for use in Pfizer Australia employee vaccination program'
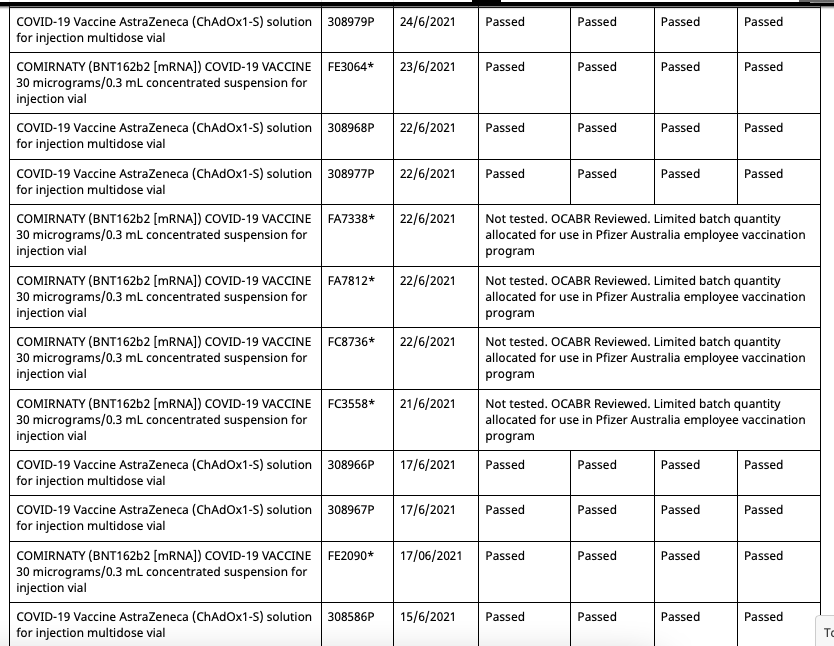
- The duplicate batch FA4598 with an associated date of 30/7/2021 created on August 11th has had its status now changed from 'Not tested, additional shipment' to 'Limited batch quantity allocated for use in Pfizer Australia employee vaccination program'
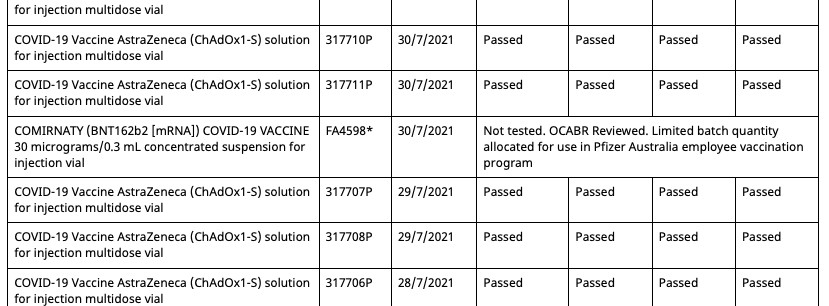
The evolution of entries of batch FA4598 alone is as follows:
Up until August 11th:
Batch FA4598 - 22 June 2021, Marked for Pfizer Employees.
From August 11th-19th:
Batch FA4598 - 22 June 2021, Marked for Pfizer Employees.
Batch FA4598 - 30 June 2021, Untested additional shipment.
From August 19th - September 21st:
Batch FA4598 - 30 June 2021, Untested additional shipment.
From September 19th - December 9th(Present):
Batch FA4598 - 30 June 2021, Marked for Pfizer Employees.
What can be made of this?
Well in summary, it has been demonstrated to be known that the mRNA gene therapy vaccine solution can be produced using two substantively different processes, a clinical process and a commercial process. It has been demonstrated to be known that commercial batches have a higher potential for risk than clinical batches. It has been demonstrated to be known that the EMA expressed concerns with Pfizer over discrepancies between the clinical and commercial batches, pertinent to the safety and efficacy of the mRNA gene therapy vaccine solution produced. It is has been demonstrated to be known that Pfizer and Pfizer corporate associates vaccinate their employees and their employees families with batches that are separate from commercial batches given to the general population. It is reasonable to believe that data provided by Untied States post market surveillance system, VAERS, may indicate an extremely uneven distribution of reported incidents of AE and death across batches of the mRNA gene therapy vaccine solution, indicating some batches are significantly more risky than others. It has been demonstrated to be known, that at least in Australia, Pfizer employee batches are not tested by the National regulatory agency which tests all commercial batches, and that batches marked for Pfizer employees in Australia underwent odd modifications as to their regulatory status and use within the country.
Based on the totality of evidence, it is reasonable to believe that specific batches of the mRNA gene therapy vaccine solution produced using commercial batch process 2 may lead to substantially higher reports of adverse events and death than batches of the mRNA gene therapy vaccine substance produced using clinical batch process 1. If this is reasonable to believe, then it would reasonably follow those aware of the increased risk of commercial batches, would seek to reduce this risk through administering a mRNA gene therapy vaccine solution from a clinical batch to themselves and families.
Whether or not this is true, I do not know.